Agrawal M, Maitin N, Rastogi K, Bhushan R. Seeing the unseen: diagnosing acromegaly in a dental setup. BMJ Case Rep. 2013; 2013 https://doi.org/10.1136/bcr-2013-200266
Roumeau S, Thevenon J, Ouchchane L Assessment of oro-dental manifestations in a series of acromegalic patients, the AcroDent study. Endocr Connect. 2020; 9:824-833 https://doi.org/10.1530/EC-20-0176
Dekkers OM, Biermasz NR, Pereira AM Mortality in acromegaly: a metaanalysis. J Clin Endocrinol Metab. 2008; 93:61-67 https://doi.org/10.1210/jc.2007-1191
Colao A, Ferone D, Marzullo P, Lombardi G. Systemic complications of acromegaly: epidemiology, pathogenesis, and management. Endocr Rev. 2004; 25:102-152 https://doi.org/10.1210/er.2002-0022
Dal J, Feldt-Rasmussen U, Andersen M Acromegaly incidence, prevalence, complications and long-term prognosis: a nationwide cohort study. Eur J Endocrinol. 2016; 175:181-190 https://doi.org/10.1530/EJE-16-0117
Rinkoff S, Adlard RE. Embryology, craniofacial growth, and development.Treasure Island, FL, USA: StatPearls Publishing; 2023
Jing J, Hinton RJ, Feng JQ. Bmpr1a signaling in cartilage development and endochondral bone formation. Vitam Horm. 2015; 99:273-291 https://doi.org/10.1016/bs.vh.2015.06.001
Dahlqvist P, Spencer R, Marques P Pseudoacromegaly: a differential diagnostic problem for acromegaly with a genetic solution. J Endocr Soc. 2017; 1:1104-1109 https://doi.org/10.1210/js.2017-00164
Reid TJ, Post KD, Bruce JN Features at diagnosis of 324 patients with acromegaly did not change from 1981 to 2006: acromegaly remains under-recognized and under-diagnosed. Clin Endocrinol (Oxf). 2010; 72:203-208 https://doi.org/10.1111/j.1365-2265.2009.03626.x
Katznelson L, Laws ER, Melmed S Acromegaly: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014; 99:3933-3951 https://doi.org/10.1210/jc.2014-2700
Acromegaly is a rare and underdiagnosed acquired disorder. It is commonly caused by an underlying pituitary adenoma that often requires surgical excision. Diagnosis is normally preceded by ~5–10 years of excessive growth owing to hypersecretion of growth hormone. This can have long-term medical complications and result in a reduced life expectancy. Acromegalic patients present to a number of healthcare professionals prior to diagnosis, including general dental practitioners. As changes in occlusion are often a patient's primary concern prior to acromegaly diagnosis, it is important that all dentists are aware of the presentation and differential diagnoses of such disorders.
CPD/Clinical Relevance: Dentists should be aware of the initial presentation of acromegaly to facilitate early diagnosis.
Article
Acromegaly is an uncommon condition manifesting in characteristic changes caused by hypersecretion of growth hormone (GH), most commonly from pituitary somatotroph cells in benign adenomas.1 These changes typically affect peripheral parts of the body, such as the hands, feet and ears, as well as altering facial features, including the jaws, forehead and nose (Table 1).
Table 1. Typical physical signs of acromegaly.
Enlargement of hands and feet
Enlargement of the nose
Frontal bossing
Growth of ears
Prognathism
Macroglossia
Thickening of lips
Change in interdental spacing
Owing to the insidious nature of this condition, any diagnosis is preceded by numerous years of growth (approximately 6–8 years), which in turn, can have lasting impacts on a patients quality of life.2,3 Diagnosis is predominantly initiated by patients presenting to healthcare professionals regarding other related symptoms.12 Dentists are often the first healthcare professionals to whom these patients will present.4,5 As there can be life-threatening complications associated with this condition, it is important to keep this disorder in mind when patients present with unusual changes in occlusion beyond the age of maturation.4,6,7
Acromegaly
Acromegaly is primarily caused by pituitary adenomas, and presents as multiple distinctive clinical changes.1,2,3,8,9,10 These adenomas most commonly arise from somatotroph cells in the anterior pituitary and are most often treated by surgical removal.1,8 This dysregulation of GH results in characteristic changes to the body, which develop over a long period of time. Often these subtle changes are not noticed by patients day to day, and only become obvious when they have changes in ring size, shoe size or their occlusion. Characteristic clinical manifestations include excessive growth of hands and feet, frontal bossing, enlargement of the nose and ears, macroglossia, thickening of the lips and mandibular prognathism.1,4,8 The hypersecretion of GH can also impact multiple other organs and systems of the body, including the pancreas, heart, and joints.8 As a result, patients are at higher risk of developing comorbidities. These include hypertension, diabetes and cardiomyopathy, as well as increased cerebrovascular and pulmonary dysfunction.1 The insidious nature of acromegaly makes this condition quite challenging to identify and diagnose early. Patients can undergo years of active change owing to hypersecretion of growth hormone prior to diagnosis. Over time, these comorbidities can significantly impact the morbidity and mortality of acromegalic patients. Some data have shown a 32% increased all-cause mortality for these patients.11 Therefore, although it is a rare condition, it is important that acromegaly is diagnosed at the earliest instance. These patients often present to several different healthcare practitioners, including dental surgeons and general practitioners, with differing symptoms. Typical complaints include change in occlusion, change in ring size or shoe size or changes to menstruation cycle.1 A successful early diagnosis of acromegaly will help to reduce the number and severity of the comorbidities, and thus help to reduce the mortality and lifelong effects.
Normal craniofacial growth
A common primary complaint of patients with undiagnosed acromegaly is altered occlusion. It is therefore important to consider how this differs from normal craniofacial development.
Craniofacial growth begins 5–7 weeks in utero with the formation of the facial processes (formed of mesenchyme bound by epithelium).12 Growth occurs using two different mechanisms: endochondral ossification and intramembranous ossification.13,14 The mandible develops from the first pharyngeal arch, and its growth and shape are aided by the Meckle's cartilage.14,15 Mandibular development initiates from bilateral areas of condensation of mesenchyme, from which point intramembranous osteogenesis continues anteriorly and then posteriorly, forming the body of the mandible.12,13
Therefore, although Meckle's cartilage does not form part of the mandible, it influences the shape of growth and development.15 The condylar cartilage grows independently to the body and ramus of the mandible by endochondral ossification, and fuses at 4 months in utero. Further growth occurs in utero from secondary growth cartilages, leading to further development of the condylar process (condylar cartilage), coronoid process (coronoid cartilage) and body (at the symphyseal cartilage).14
Growth continues throughout childhood and puberty with the main site of endochondral ossification being at the condylar cartilage. Intramembranous ossification remodels the mandible, causing resorption on the anterior surface of the ramus, and apposition on the posterior surface of the ramus, chin, coronoid and alveolar ridges. This results in forward and downward growth away from the cranial base.14 The majority of mandibular growth and changes occur between the ages of 14 to 16 years for females, and 16 to 18 years in males. It can continue up until the age of 20, at which point mandibular growth usually ceases and the occlusion stabilizes.16,17 However, growth often continues up to the age of 20, at which point it usually stabilizes.
It is therefore important to take note when patients attend complaining of a changing occlusion beyond this age, and consider acromegaly as a possible diagnosis.
Differential diagnoses
Despite the described facial and acral overgrowth symptoms being typical of acromegaly, there are several differential diagnoses that could be considered.
Gigantism
Gigantism and acromegaly often have a similar aetiology (a pituitary adenoma causing oversecretion of growth hormone), and the terms are commonly used interchangeably; however, gigantism occurs before the epiphyseal plates of the bones have fused during childhood. Therefore, the symptoms present during this time. Typical symptoms include delayed puberty, prognathic jaw, enlarged tongue, spreading of teeth, frontal bossing and prominent enlargement of feet and hands.18 As described above, patients with acromegaly present with very similar symptoms, but during adulthood.
Sotos syndrome
Sotos syndrome is a condition most commonly caused by de novo mutations, and patients show symptoms from a very young age.19 It is rare and patients present with acromegalic symptoms, but testing shows no excessive secretion of growth hormone. Genetic testing is required to aid diagnosis of this congenital syndrome.
Marfan syndrome
Marfan syndrome is another genetic disorder that results in selective overgrowth. This condition affects the connective tissues in the body, impacting a variety of systems, including the cardiovascular and skeletal. Among other more severe symptoms, patients with Marfan syndrome often show signs of overgrowth of the long bones in the arms and legs, long fingers, a high arched palate and crowded teeth.20,21 This condition is primarily due to a mutation in the fibrillin-1 gene, causing an increase in transforming growth factor beta. Often a genetic test can assist the diagnosis.
Patients with this genetic condition typically present with thickening of the skin of the face (pachyderma), clubbing of the fingers and toes and excessive sweating.20 As some of these symptoms are similar to those seen in acromegalic patients, it should be ruled out as a differential diagnosis.
Case presentation
A 40-year-old male was referred by their general dental practitioner to the orthodontic department at Pinderfields General Hospital regarding a change in occlusion. On attending, the patient complained of an 18–24-month history of change in ‘bite’; the patient felt that his mandible had grown. The patients' medical history revealed that he had von Willebrand disease.
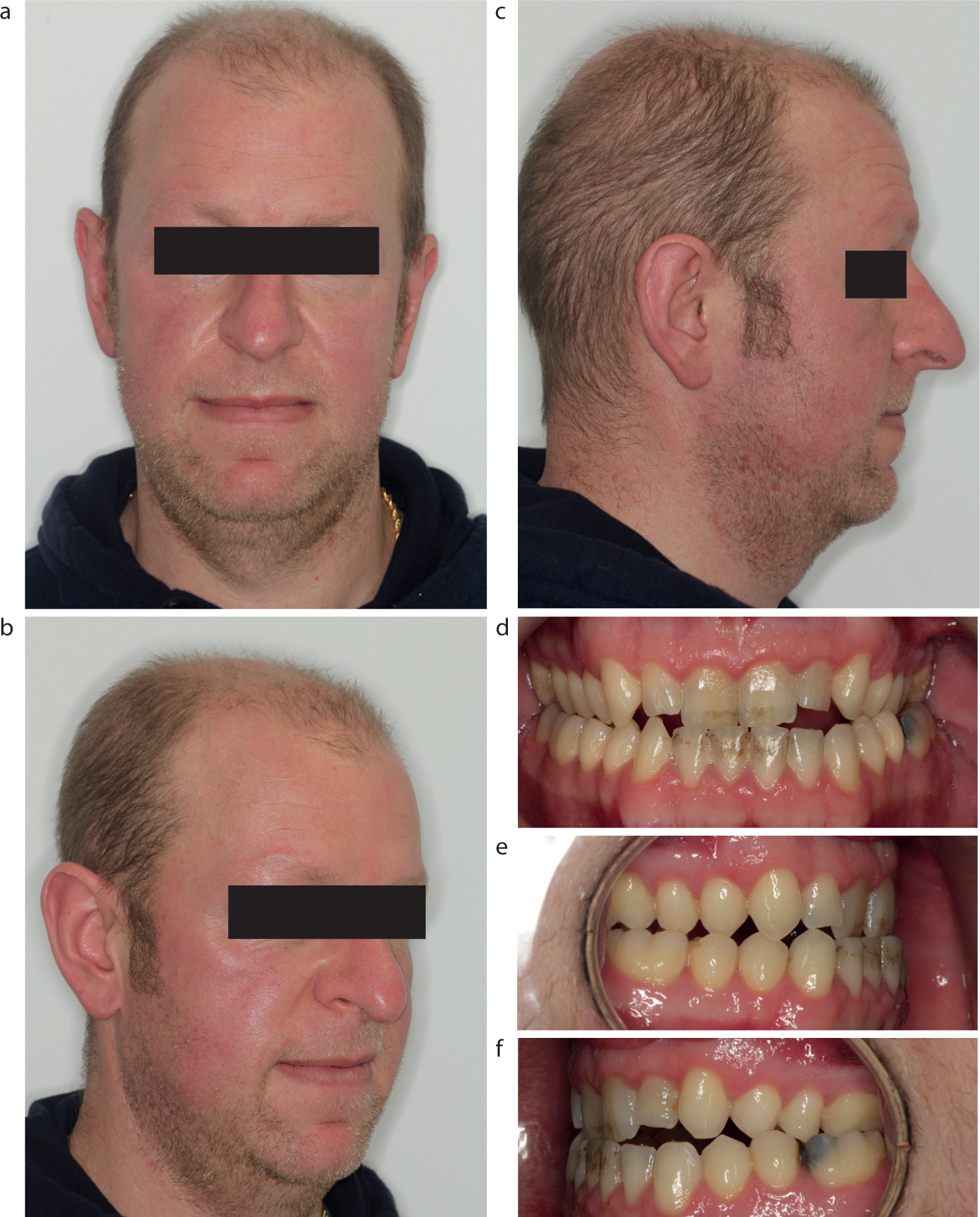
Extra-oral examination of the patient showed a widened nose, frontal bossing (Figure 1) and large hands with thickening of the skin on the dorsal aspect. The patient exhibited a Class III incisor relationship, with a reverse overjet of -1.5 mm, on a skeletal base 1 with increased vertical proportions.
Figure 1. (a-f) Initial presentation records taken at first orthodontic appointment.
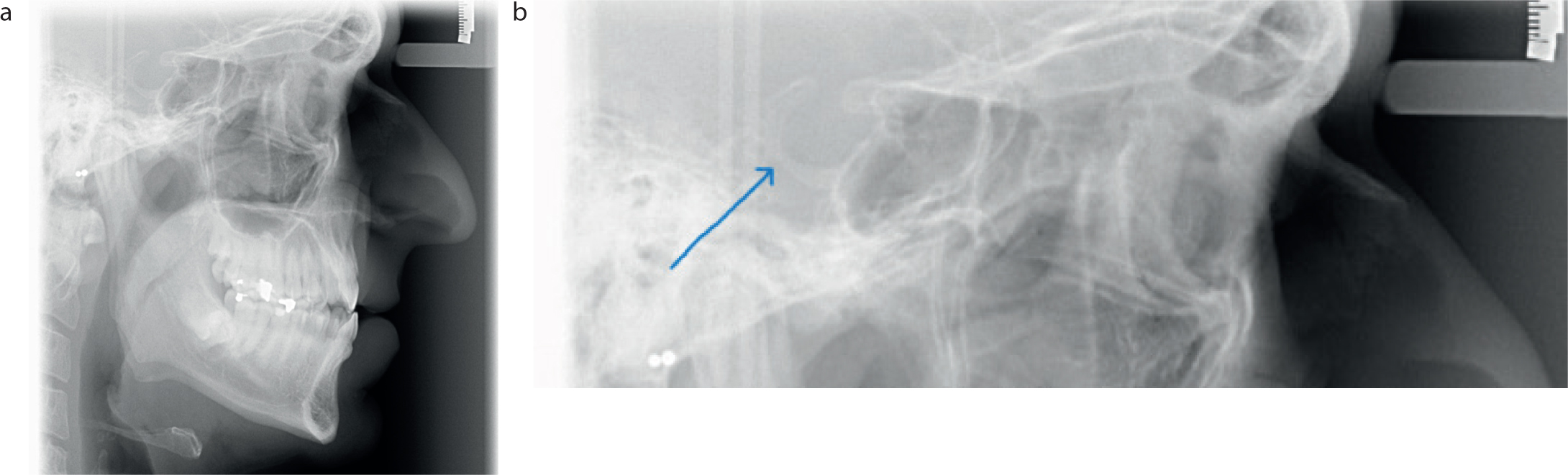
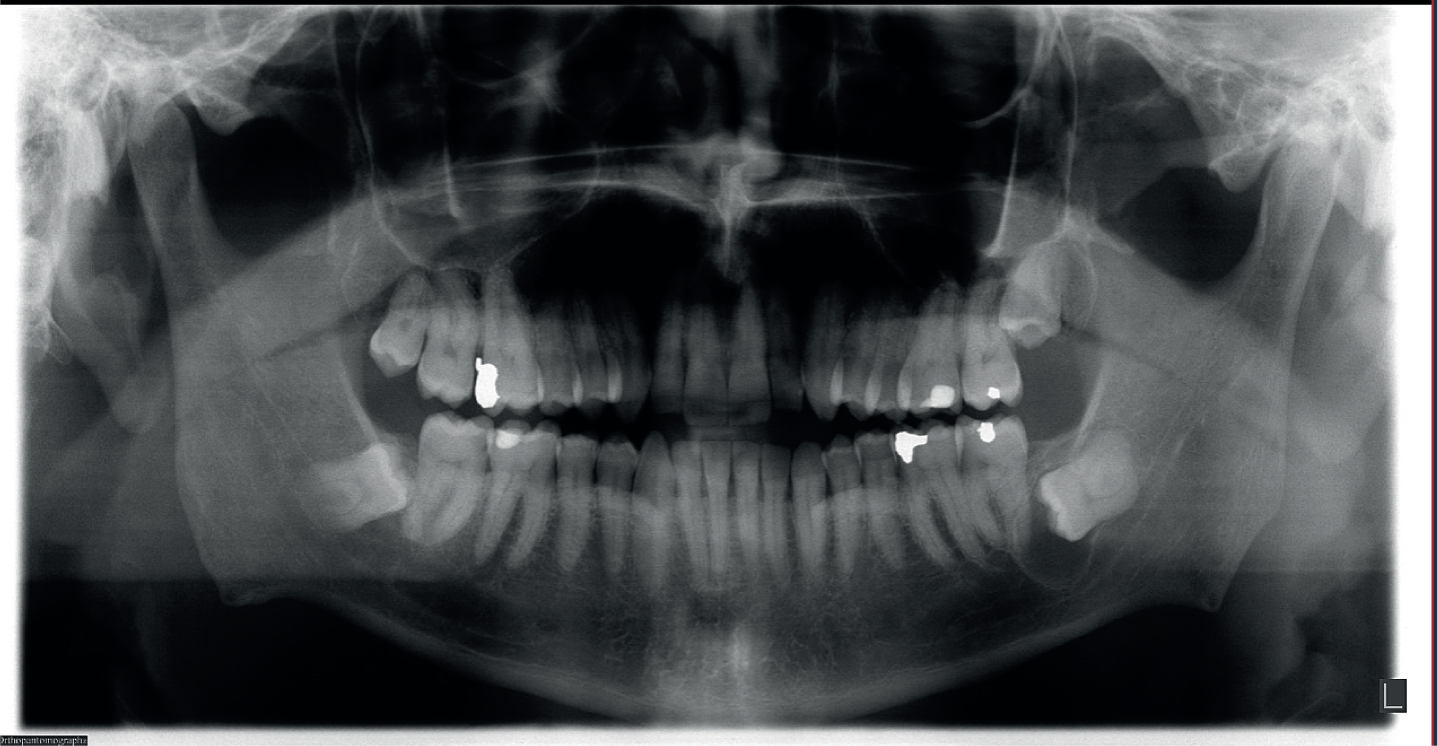
Owing to the patient's history and presentation, a provisional diagnosis of acromegaly was made, and radiographic investigations were undertaken. An orthopantomogram (OPG) and cephalogram were requested (Figures 2 and 3). The OPG showed some symmetrical lengthening of the condyles and prominent antegonal notches. There was also an incidental finding of a possible dentigerous cyst associated with the lower left third molar. The lateral cephalogram showed an enlarged sella turcica and showed Class III incisors on a mild Class 3 skeletal base.
Figure 2. Lateral cephalograms (a) taken at first orthodontic appointment and (b) highlighting enlarged sella turcica.Figure 3. Orthopantomogram taken at first orthodontic appointment.
During this visit, records were taken, including photographs and impressions, for study models. Further haematological investigations were requested, as discussed below, to confirm the suspected diagnosis of acromegaly and a joint review appointment was organized with consultants in orthodontics and oral and maxillofacial surgery.
Haematological investigations
Initial haematological investigations, including insulin growth factor type 1 (IGF 1), growth hormone and prolactin were requested. These showed a significantly increased IGF-1 of 86.7 nmol/L (normal range 12.9–30.0 nmol/L); however, both prolactin (116 µg/L) and growth hormone (3.8 ng/mL) were within normal range. At this point the patient was referred to the endocrinology department (Table 2).
Table 2. Initial and diagnostic haematological investigations required for diagnosing acromegaly.
Investigation
Test
Initial
Insulin growth factor type 1 (IGF 1)
Growth hormone (GH)
Prolactin
Diagnostic
Growth hormone measurements under oral glucose tolerance test (OGTT)
Endocrinology assessment
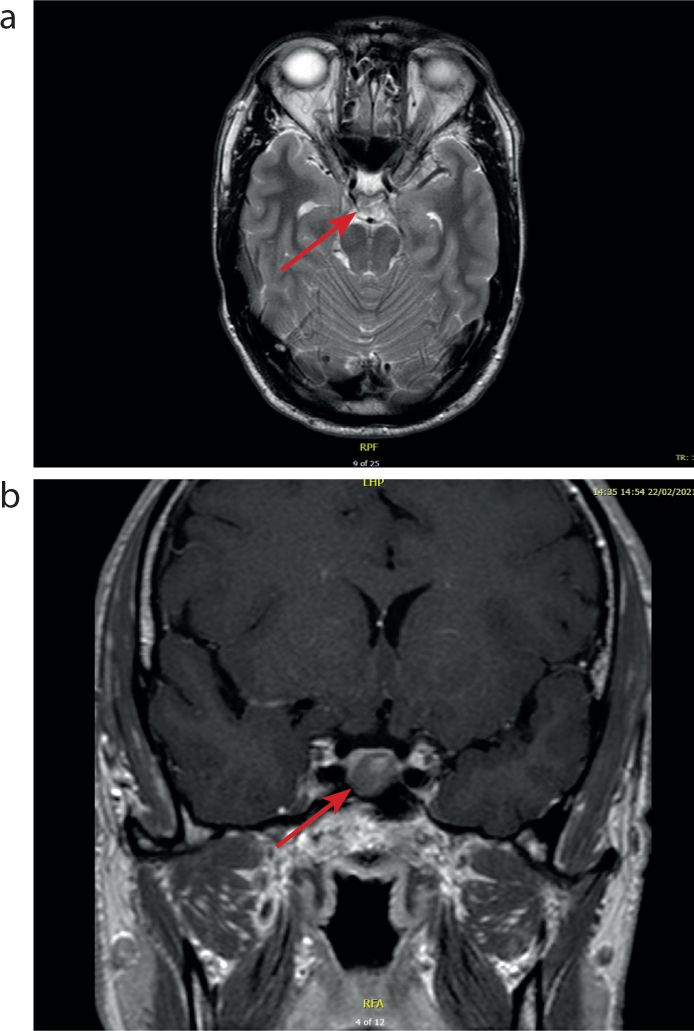
During this assessment it was noted that there were no abnormalities in visual fields; however, there was a moderate sized multi-nodular goitre. Further investigations were requested, including an MRI pituitary scan and measurement of growth hormone under oral glucose tolerance test (OGTT), the gold standard for confirmation of the diagnosis of acromegaly. The MRI scan showed a well-defined central/left paracentral poorly enhancing lesion measuring 11 x 12x 10 mm (w x h x l) (Figure 4). This appeared to be compatible with a macroadenoma of the pituitary, with no significant supra-cellular extension and no deviation of the pituitary stalk. The optic chiasm and the optic nerve appeared normal, and there appeared to be no involvement of the cavernous sinus. The growth hormone levels measured during the OGTT demonstrated continued GH elevation that was not suppressed. These results are consistent with a diagnosis of acromegaly as a result of a pituitary macroadenoma.
Figure 4. MRIs showing (a) an axial and (b) coronal view of the pituitary macroadenoma.
Treatment
The patient was referred to Leeds General Infirmary for surgical removal, through a transsphenoidal approach, of the pituitary adenoma, with immediate and long-term review undertaken by the endocrinology department at Pinderfields General Hospital.
Discussion
Acromegaly is a rare disorder, with a mean annual incidence of only three cases per 1 million persons.2 Although awareness of this disorder has been increasing, it has been shown that it continues to be underdiagnosed, with an assumed delay of 5–10 years of disease progression prior to diagnosis.11,22 This has remained consistent for 20 years, as the age at diagnosis has not reduced, indicating the need for increased awareness in healthcare professionals.11 As dental malocclusion, a result of excessive mandibular growth, is one of the reported primary complaints of these patients, it is important for dentists and orthodontists to have an understanding of this disorder and be able to refer appropriately to endocrinology where required.1,4,6,9 It has been demonstrated that often these patients present to a number of different healthcare professionals with seemingly unrelated problems prior to their subsequent diagnosis of acromegaly.4,22 It is particularly important that these cases are recognised and diagnosed promptly because there can be numerous long-term systemic complications that can impact a patient's quality of life. These include diabetes mellitus, heart failure, venous thrombo-embolism, sleep apnoea and arthropathy.2,4,7,11 Furthermore, these long-term complications and the continued hypersecretion of growth hormone can reduce a patient's life span.4,8
For confirmation of diagnosis, biochemistry is required. An increased serum growth hormone concentration that is not suppressed following an oral glucose tolerance test, and an increase in serum concentration of IGF-1 are the gold standard markers and without which a diagnosis cannot be confirmed.10
As in this case, pituitary tumours are most commonly the cause of these acromegalic changes and the treatment of choice is often surgery.4,6,7,23 However, medical and radiotherapeutic methods are also available options.